
お客様のご意見・ご要望のご紹介

奈良 雅之 先生
東京医科歯科大学教養部化学 (ご所属・役職は2007年4月発行時)
緒言
赤外分光法の生体分子への応用は、かつて分散型赤外分光光度計しかなかった時代にはタンパク質、ポリペプチド、脂質などの固体試料の測定が精力的に行われていたが、水溶液中での生体分子の解析は、水の強い吸収による妨害を受けるため測定できないことが多かった。そのために、タンパク質の測定は重水溶液もしくは固体フィルムにする必要があった。フーリエ変換型分光(FTIR)計が主流になった今日においては、この問題は解決済みということになっている。すなわち、水の吸収の影響を受けるタンパク質アミドIバンドの解析は、試料の透過光路を10 μmまでに狭くすれば、溶液のスペクトルと溶媒のスペクトルの差演算を施すことにより、構造解析できるはずである。しかしタンパク質のFTIRが必ずしも一般的に測定されていない理由の一つとして、水蒸気の除去という基本的な操作が意外と難しく、再現性のある質の高いデータを得るために熟練が必要である点が挙げられる。FTIRによるタンパク質の構造研究は、大きく主鎖構造と側鎖構造に分けられるが、アミドIバンドの解析を中心とした二次構造解析がよく知られており、総説1-3)や論文がこれまでに数多く報告されている。このアミドIバンドは水蒸気並びに水のバンドと重なる領域であるため、水蒸気の除去や溶媒の差し引きが不完全であるとスペクトルにアーティファクトなバンドが現れる恐れがある。他方、側鎖の構造解析に関してはタンパク質の機能によって着目するバンドが異なり、例えば側鎖の帰属はBarthらの総説にまとめられている4)。今回FTIR TALK LETTERにこれまでの研究を寄稿するに際して、タンパク質水溶液のスペクトルを得るための基本的な事項も合わせて述べておきたい。
■タンパク質のFTIRスペクトルを得るための水蒸気除去、水の引き方
低分子化合物の固体サンプル、有機溶媒などの測定では、分光器のパージなどをそれほど気にしなくても満足できるスペクトルが得られる。しかし、タンパク質水溶液のスペクトルでの基本的な注意点は、光路上の水蒸気の除去と溶媒を差し引く演算処理である。これらの操作をいい加減に行うと、データの解析、特に二次微分演算処理でスペクトルの質を落としてしまう。
<水蒸気除去>
乾燥空気供給装置が備わっている分光器であれば、実験前日からパージを始めれば、分光器内の水蒸気はかなり除去できる。サンプル部とリファレンス部を備えたデュアルビームの装置であれば、水蒸気のことをそれほど気にしなくても水蒸気の影響の少ないスペクトルが得られる。しかし、乾燥空気供給装置がない場合、たいていは窒素ガスでパージすることになり、窒素ボンベを消耗品として使うので、測定の度に前日からパージするわけにはいかない。さらに分光器がシングルビームタイプであれば測定時間がずれることから水蒸気は後から差し引くことになる。分光器メーカーによってはデフォルトとして水蒸気除去プログラムが入っているところもある。実際にこの除去プログラムを使うと、一見スペクトルから水蒸気が消えているように見える。赤外スペクトルに特に演算処理を施さないのであれば、このプログラムを活用したほうが見映えみは良くなる。しかし、タンパク質の主鎖ならび側鎖の構造解析においては、このようなプログラムはあまり役に立たないどころか、かえってアーティファクトを生んでしまう原因になる。水蒸気はそのときの温度、湿度などの環境で変化するので、水蒸気のバンドを消すためには、その日に測定した水蒸気のスペクトルを用いることが望ましい。実際に、数時間以内で測定した水蒸気であれば、かなり水蒸気を除去することができる。最終的には、サンプルによるバンドがない2000~1700 cm-1の領域で水蒸気のシャープなバンドが出ていないことで確認できる。最近は、マクロの透過型測定よりもATR測定や顕微赤外測定の方がかえって簡便になってきたが、分光器にATRユニットや顕微赤外ユニットを外づけのアクセサリーとして用いると、測定はシングルビームになる。しかもアクセサリーは意外にパージに関する配慮が十分になされていないことが多いので、水蒸気に関しては注意が必要である。
<水の差し引き>
重水の場合はDOD変角振動が1200 cm-1付近に現れるので、アミドI領域(1700-1600 cm-1)を解析するのにバンドのオーバーラップを気にする必要はない。しかし、軽水の場合は、水のHOH変角振動とタンパク質のアミドIバンドが重なるので、水を引くときは2300~2000 cm-1の水の結合音と考えられるブロードなバンドが平らになるように差をとるのが一般的である。水の差し引きを円滑に行うためには、試料のサンプルの厚みをスペーサーで15 μm以下にすることである。20 μmの厚さで水の吸光度は2近くになるので1650 cm-1付近のバンドが飽和してしまう。透過法を用いずにATR法を用いると、水の吸収を低く抑えることができるので、タンパク質水溶液の測定には有効である。
今回紹介するカルシウム結合タンパク質の側鎖COO-基の研究では、COO-逆対称伸縮振動バンドを解析するために、アミドⅡバンドとの重なり避けることが必要となり、重水溶液での測定が必要になる。タンパク質主鎖のNHはすべてD化し、重水溶液試料を準備した。タンパク質濃度は1~2 mMに調製した。測定はすべて透過型(マクロ)で行った。
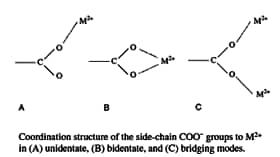
■アカザラガイトロポニンC(天然型)の金属イオンとの特異的相互作用5)
トロポニンC(TnC)は筋収縮を制御するCa2+結合タンパク質である6)。脊椎動物の骨格筋や心筋では、細胞内のCa2+濃度に応じて、TnCは最大4つのCa2+を結合し、立体構造を変化させることにより、筋収縮のonとoffを制御している。一方,無脊椎動物のアカザラガイ閉殻筋TnCは、脊椎動物のTnC同様、4つのEFハンドモチーフ(helix-loop-helix)7)をもつにもかかわらず、1つしかCa2+を結合しない8)。このカルシウム結合部位は最もC端側のEFハンドモチーフ(サイトⅣ)に位置する。そこで、アカザラガイトロポニンC並びにその変異体のCa2+,Mg2+配位構造についてFTIRを用いて解析した。赤外COO-逆対称伸縮振動バンドは金属イオンに結合するグルタミン酸(Glu, E)もしくはアスパラギン酸(Asp, D)側鎖COO-基の配位様式についての情報を与え、その波数に基づいて,unidentate型,bidentate 型,birdging 型などの配位様式(図1)を識別することができる9), 10)。bridging型の2つの金属イオンのうち一つが水分子などに置き換わる場合をpseudo-bridging型といい、タンパク質水溶液中ではunidentate型は実際にはpseudo-bridging型になっていると考えられる11)。
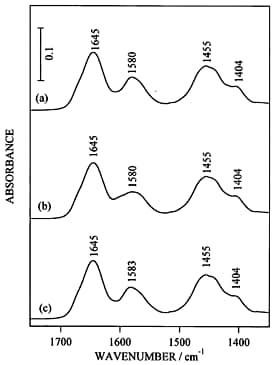
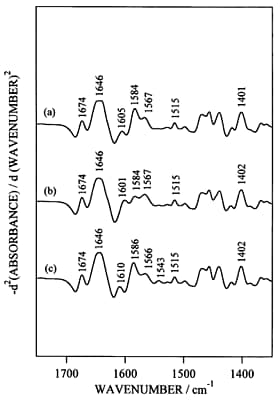
通常の赤外吸収スペクトルだけでは金属イオンの結合による構造変化を比較することは難しいので、二次微分演算あるいはフーリエセルフデコンボルーションなどの演算処理を施す必要がある。図2にapo型(二価金属イオンフリー型),Mg2+結合型,Ca2+結合型の赤外吸収スペクトル、図3に二次微分スペクトルを示す。赤外吸収スペクトルだけを眺めてもよく似ているということぐらいしか議論できない。しかし、二次微分演算を施すと、側鎖に関する詳しい議論ができるようになる。図3においてCa2+結合型のみで1543 cm-1にバンドが現れたが,これはGlu142の側鎖COO-基がCa2+と二座配位型で結合したものと解釈した。一方,Mg2+結合型の1600 cm-1及びCa2+結合型の1586 cm-1のバンド強度の増大は,Asp131とAsp133の側鎖COO-基がMg2+およびCa2+とpseudo-bridging型で結合したことを示唆した。生理的条件下では、offではMg2+がonではCa2+がトロポニンCと結合する可能性が考えられるので、on/offの機構として図4に示す配位構造のモデルが考えられる。蛇足ではあるが、もしも水蒸気の引き残りがあると、二次微分演算で水蒸気のピークがエンハンスされて、タンパク質の情報がかき消されることになったであろう。
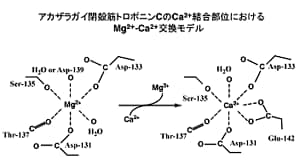
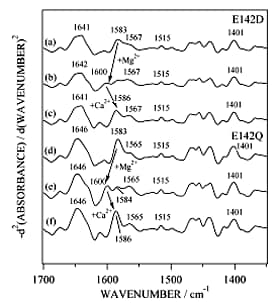
■ E142Q変異体のMg2+,Ca2+配位構造12), 13)
結合サイトIVの12番目のグルタミン酸をグルタミン(Gln, Q)で置き換えた変異体をE142Qと呼ぶことにする。E142Qの赤外二次微分スペクトルを調べると、Mg2+負荷状態で1600 cm-1のバンド強度が増加したことにより、Asp131, Asp133の側鎖COO-基はpseudo-bridging型でMg2+と結合したものと考えられる。この結果は、これまで天然型のMg2+結合に伴うスペクトル変化の結果とほぼ一致した。そこで、Mg2+滴定を行って1600 cm-1のバンドの強度変化を調べたところ、Mg2+の親和性は天然型とE142Qでほぼ同じであることがわかった。したがって,天然型のGlu142側鎖COO-基はMg2+配位に関与していないことが結論づけられた。また、E142QのMg2+結合の影響についてCDや蛍光スペクトルでも調べたところ、天然型と同じ挙動を示した。一方、Ca2+負荷による影響は、12位の側鎖COO-基がなくてもAsp131とAsp133の側鎖COO-はCa2+と結合することがわかった(図5d-f)。しかし、CDや蛍光スペクトルを測定するとCa2+結合の効果は天然型のものとは異なり、Glu142側鎖COO-基がアカザラガイトロポニンCの活性化のon/off機構に直接関わっていることが示唆された。E142D変異体に関しても同様にCa2+と結合としても1543 cm-1にはバンドが現れなかったが、Mg2+結合状態で観測される1600 cm-1のバンドがCa2+負荷により消えたことにより、結合サイトIVにCa2+が入ったことが示された(図5 a-c)。
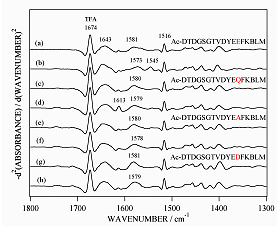
■ 合成ペプチドアナログによるアプローチ13) ,14)
タンパク質の側鎖の帰属には、アミノ酸やペプチドなどのモデル化合物のスペクトルと比較するのが常套手段である。ウサギ骨格筋トロポニンCのサイトⅢに関してNMRによる報告15)が1980年代にされていたので、まずこのペプチドに焦点を当てて解析を行った。その結果、カルシウム結合部位に相当するペプチドはCa2+と結合せず、COO-基の二座配位のバンドが観測されるためにはカルシウム結合サイト12残基の他に少なくともC端側に5残基のばしたもの、すなわち17残基の鎖長が必要であることがわかった14)。そこで、アカザラガイトロポニンCのサイトIVについても17残基の合成ペプチドアナログを用いて赤外測定を行った。結合部位の12位のグルタミン酸を他のアミノ酸で置き換えたペプチドを用いて、COO-逆対称伸縮振動領域を調べたところ、天然型のモデルペプチドでは1545 cm-1付近にバンドが観測されたが、置換型のモデルペプチドでは観測されなかった(図6)。この結果、これまでにアカザラガイトロポニンCで帰属した1543 cm-1のバンドは142位のグルタミン酸がCa2+と二座配位型で結合したことに由来することを裏付けることができた。また、グルタミン酸をアスパラギン酸で置換したE142DペプチドでもCOO-逆対称伸縮振動バンドの低波数シフトが観測されなかったということは、側鎖のメチレン基が1つ欠けただけでも配位構造に重大な影響を与えることを示唆するものであり、タンパク質の構造機能相関を理解するために興味深いデータと考えられる。
■結語
カルシウム結合タンパク質と金属イオンとの特異的相互作用を調べる方法として、FTIRが有用なアプローチであることを紹介した。特に変異体やモデルペプチドを利用することにより,カルシウム結合タンパク質の活性化のon/off機構を直接的に解明できることが期待される。
タンパク質構造に関してX線結晶構造解析や多次元NMRで得られる情報量に比べればFTIRで得られる情報量ははるかに少ない。しかし、構造と機能との相関に関わる情報がピンポイントで得られるという点がFTIRの特徴と考えられる。今後FTIRが活躍しそうな研究テーマとしてintactな状態でのタンパク質構造解析が挙げられる。不溶性タンパク質や不均一な生体システムはX線結晶構造解析やNMRといった構造生物学の常套手段が苦手とする研究対象である。それに対してFTIRは基本的にサンプルの状態によらず測定可能である。例えば、ウシ卵子の表層部に存在する透明帯タンパク質を非破壊的に測定できる手段としてATR/FTIRが有用であることを明らかにした16)。このように生物システムのintactなタンパク質に焦点を当てていくと、FTIRの有用性はさらに高まるものと期待できる。
■謝辞
アカザラガイトロポニンCのFTIRに関する研究は、東京大学大学院農学系研究科の田之倉優教授の協力でここまで発展してきた。タンパク質の精製、調製では湯本史明博士の協力を得た。また、カルシウム結合部位の合成ペプチドアナログは産業技術総合研究所の森井尚之博士に提供していただいた。
文献
r
