
お客様のご意見・ご要望のご紹介

栗山 翔吾 先生
東京大学大学院工学系研究科応用化学専攻 助教 (ご所属・役職は2022年8月発行時)
アンモニアは,生体を構成するタンパク質やDNA の窒素源となる化学物質で,種々の含窒素化成品の原料として用いられている。近年では,アンモニアが容易に液化し,高いエネルギー密度を有し,かつ燃焼しても二酸化炭素を排出しないことから,脱炭素燃料の候補としても着目されている。アンモニアは大気中に約80%含まれる窒素分子から合成するが,窒素分子は非常に強い窒素‒窒素三重結合を有するため,窒素分子をアンモニアへと還元する反応,窒素固定反応は非常に困難な化学反応である。
工業的な窒素固定法は100年前に開発されたハーバー・ボッシュ法が用いられている(図1a)。ハーバー・ボッシュ法では,鉄系固体触媒を用いて窒素ガスと水素ガスを高温・高圧で反応させることでアンモニアを合成している。この反応で用いられる水素ガスは化石燃料の改質によって得ており,その製造は二酸化炭素排出を伴うとともに多大なエネルギーが必要である。そのため,化石資源に依存しない温和な反応条件下で進行するアンモニア合成法が次世代型窒素固定反応として望まれている。
一方で自然界では,窒素固定酵素であるニトロゲナーゼが大気中の常圧の窒素分子を常温でアンモニアへと変換している(図1b)。ニトロゲナーゼの活性部位は中心に炭素原子を持つ硫黄架橋鉄・モリブデンクラスター構造であることが明らかとなっている。そこで,ニトロゲナーゼの活性中心を模倣した,遷移金属中心に窒素分子が配位した窒素錯体を用いた温和な条件下での窒素固定反応の開発が古くからなされてきた。遷移金属錯体を用いた触媒的アンモニア生成反応は2003年にSchrockらによって初めて報告され,モリブデン窒素錯体が触媒として用いられた[1]。これらの例以降,いくつかの研究グループによって様々な遷移金属触媒を用いた触媒的アンモニア生成反応が達成されている[2]。
我々の研究室は3座配位子であるピンサー配位子を持つモリブデン窒素錯体を用いた触媒的アンモニア生成反応を報告している(図1c)[3,4]。この報告以降,モリブデン錯体を中心に様々な遷移金属窒素錯体を用いた触媒的窒素固定反応を報告している[5]。本稿では,最近の鉄やコバルトといった卑金属窒素錯体を用いた触媒的窒素固定反応について紹介したい[6,7]。
さて,窒素分子が金属に配位するとき,通常のσ供与に加え,金属の被占有d軌道から窒素分子のπ*軌道への逆供与によって結合次数が低下する(図1d)。遊離の窒素分子の伸縮振動は2331 cm-1 に観測されるが,配位により振動数が低波数側にシフトする。遷移金属錯体上の末端配位窒素分子の伸縮振動は赤外活性であり,その赤外吸収を測定することでその活性化度を定量化することが可能である。さらに,赤外スペクトルにおける伸縮振動の吸収パターンによって配位窒素分子の数や配位形式を知ることも可能である。このように遷移金属窒素錯体の合成において赤外スペクトルは不可欠な分析であるが,遷移金属窒素錯体はしばしば空気や水に不安定という問題点がある。赤外スペクトルの測定はKBr錠剤や有機溶媒の溶液状態で行うことが多いが,空気・水との接触を避けて調製・測定することは困難を伴う。そこでグローブボックス内に設置したIRSpiritを使用することで,こうした問題を気にせずに反応性の高い錯体の赤外スペクトルを簡便に測定できるようになった。
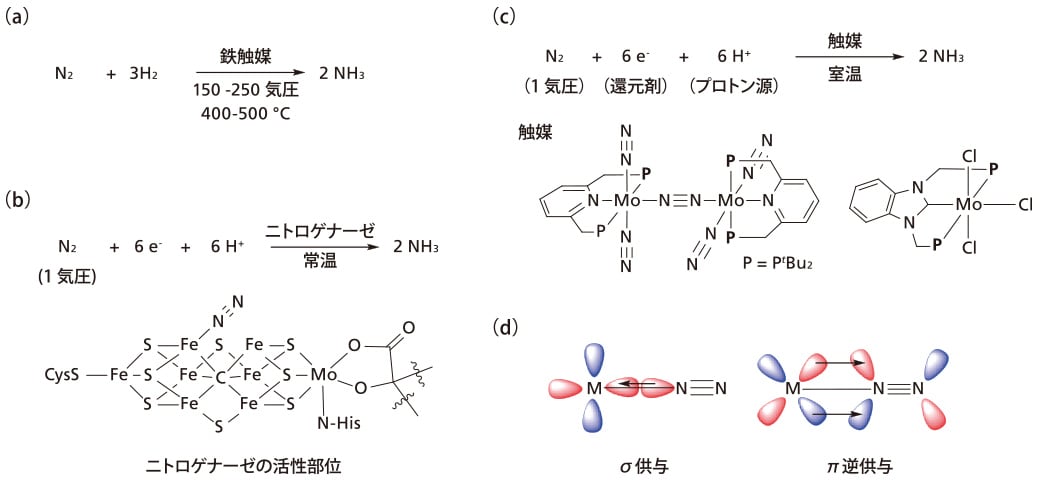
図1 (a)ハーバー・ボッシュ法 (b)ニトロゲナーゼ (c)遷移金属錯体による窒素固定反応 (d)窒素分子の配位
ニトロゲナーゼの活性部位の構造は鉄 - 炭素結合を含むクラスター構造であることが明らかになっているがその詳細な反応機構は明らかになっていない。最近の研究により,炭素原子が配位した鉄中心に窒素分子が配位していることが示唆された(図1b)。そのため,ニトロゲナーゼのモデルとして,鉄 - 炭素結合を有する鉄窒素錯体の反応性に興味が持たれる。この背景の下我々は,強い電子供与性を有し金属に強固に配位できるベンゼン骨格を含むアニオン性PCP型ピンサー配位子を持つ鉄錯体を触媒として着想した[6]。
図2a に示すPCP型ピンサー配位子を持つ鉄(I)窒素錯体(1)を合成した。錯体1の赤外スペクトルでは末端窒素配位子に帰属できる吸収が1954 cm-1 に観測された。次にこの鉄錯体を用いた触媒的な窒素分子の還元反応を検討した。触媒量の錯体1存在下,常圧の窒素ガスを還元剤としてカリウムグラファイト(KC8),プロトン源としてオキソニウム塩([H(OEt2)2]BArF4, ArF = 3,5-(CF3)2C6H3)とジエチルエーテル中-78 ℃で反応させることで触媒当たり252当量のアンモニアと68当量のヒドラジンが生成した。この反応で固定された窒素原子は触媒当たり388当量に達した。この値は従来の鉄触媒の活性より高く,本錯体が窒素固定反応に対して最も高活性な鉄触媒として働くことが分かった。
反応機構に関する知見を得るため,鉄錯体の還元を検討した(図2b)。錯体1 を窒素雰囲気下でカリウムを用いて1電子還元すると,原料の吸収に加え末端窒素配位子に帰属される吸収が1981 cm-1と1894 cm-1 に新たに2本観測され,2分子の窒素分子が鉄中心にcisで配位した錯体の生成が示唆された(図2c)。最終的にこの錯体の構造は単結晶X線構造解析により,アニオン性鉄(0)窒素錯体(2)であることを明らかにしている。このアニオン性錯体も触媒として働いたことから,アンモニア・ヒドラジン生成反応においても同様の鉄(0)窒素錯体が活性種として働いていると考えられる。
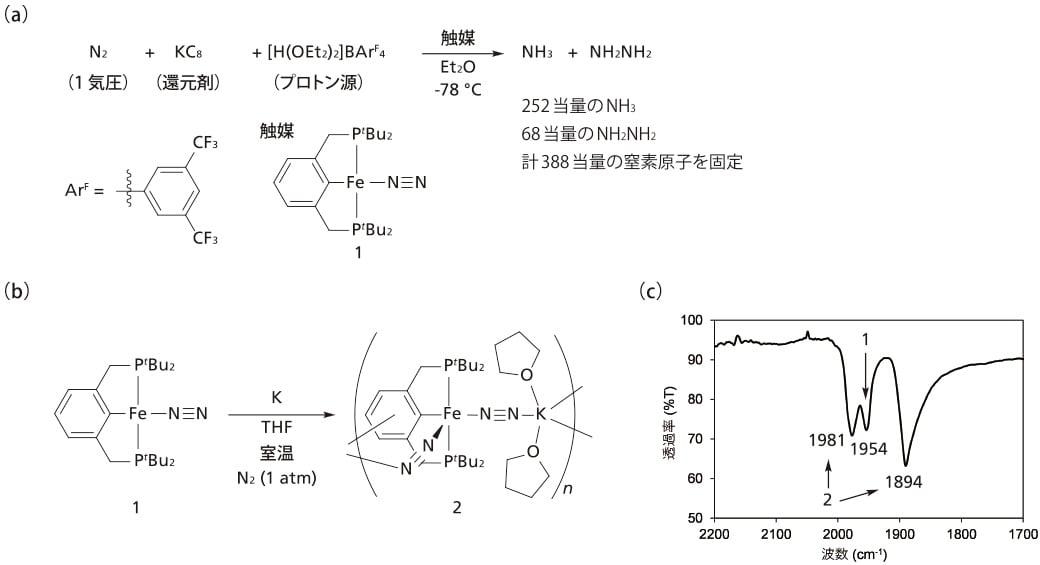
図2 (a)鉄窒素錯体を用いた窒素分子からのアンモニア・ヒドラジン生成反応
(b)鉄窒素錯体の還元反応 (c)鉄窒素錯体のIRスペクトル
シリルアミン(N(SiMe3)3)は加水分解によって容易にアンモニアへと変換されることから,窒素分子からのシリルアミン合成はアンモニア合成反応のモデル反応および窒素固定反応として広く研究されている。これまでに様々な遷移金属錯体が触媒的シリルアミン生成反応の触媒として働くことが報告されているが,中でもコバルト錯体が高い活性を有することが知られている[8]。
当研究室ではアニオン性ピンサー配位子を持つ9 族ロジウム・イリジウム錯体がシリルアミン生成反応の触媒として働くことを報告している[9,10]。そこでアニオン性ピンサー配位子を持つ同族のコバルト錯体も同様にシリルアミン生成反応の触媒として働くと考えた[7]。
PCP型ピンサー配位子を持つコバルト窒素錯体を合成した。さらに,配位子のベンゼン環に置換基を導入した錯体を合成し,置換基の電子的性質が錯体に与える影響を調べることとした。無置換の錯体3の赤外スペクトルでは,2009 cm-1 に配位窒素分子の吸収が観測される。電子供与性基であるメトキシ(OMe)基およびtert- ブチル(tBu)基を持つ錯体4と5では2005,2006 cm-1 に低波数シフトし,コバルト中心から配位窒素分子への逆供与が増加して窒素分子がより活性化されたことが示唆された。一方,電子求引性基である3,5- ビストリフルオロメチルフェニル(ArF)基を導入した錯体6では2014 cm-1と高波数シフトし逆供与が低下したことが分かった。
続いてこれら錯体を用いたシリルアミン生成反応を検討した。触媒として無置換の錯体3を用いて常圧の窒素ガスを,還元剤としてカリウムグラファイト,シリル化剤としてトリメチルクロロシランとTHF中室温で96時間反応させると,触媒当たり351当量のシリルアミンが生成した。この触媒活性はこれまで報告された他の触媒の活性より高い。次に電子供与性基を有する錯体4と5を触媒としたときは,332および371当量のシリルアミンが得られ,無置換の錯体3とほぼ同等の触媒活性が見られた。一方で,電子求引性基を有する錯体6では,生成したシリルアミンは106当量へと低下した。電子求引性基の導入で配位窒素分子への逆供与が弱くなり窒素分子が活性化されていないことが,触媒活性が低下した原因と考えられる。以上,新規に合成したコバルト錯体がこれまでで最も高活性なシリルアミン生成触媒として働くことがわかった。
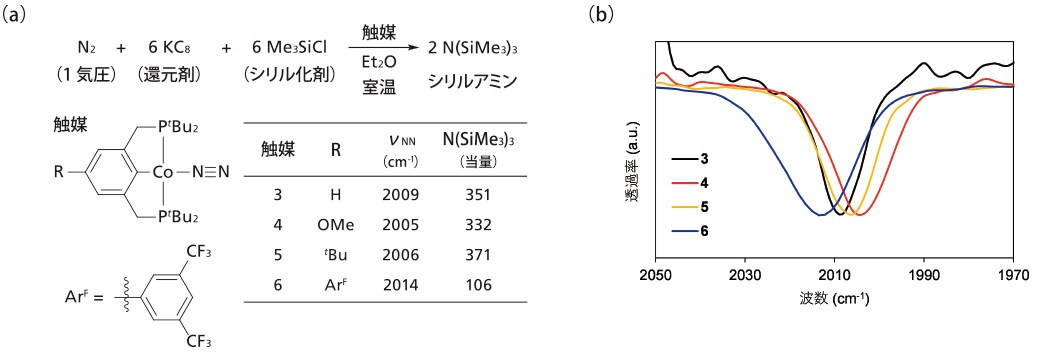
図3 (a)コバルト窒素錯体を用いた窒素分子からのシリルアミン生成反応 (b)コバルト窒素錯体のIRスペクトル
本研究ではアニオン性PCP型ピンサー配位子を持つ卑金属錯体を用いることで温和な条件下での触媒的窒素固定反応が効率的に進行することを見出した。現在はより高活性な触媒開発や,電気化学反応などを合わせてより環境調和型の反応系開発に取り組んでいる。
謝辞
上記の研究は西林仁昭教授の下で得られた成果であり,研究室構成員の方々のご協力に感謝申し上げます。共同研究者である九州大学吉澤一成教授,大同大学田中宏昌教授の研究グループにもこの場を借りて感謝申し上げます。
