試料溶媒中の溶存空気由来のピークについて
HPLC分析では,しばしば出所不明のピーク(ゴーストピーク)が出現し,悩まされますね。 特に医薬品の不純物試験をしている場合は,やっかいなものです。ゴーストピークの原因はさまざまですが,案外気づかれない原因の一つに,試料溶液中の溶存空気によると考えられるものがあります。今回は逆相分離/UV検出でのお話です。
1. 溶存空気(酸素)由来らしきピークの出現
さて,試料溶液と移動相との間で有機溶媒の種類や組成が違えば,何らかのピークが出ると予想出来ます。ところが,移動相そのものを試料溶液として注入してもピークが出ることがあります。しかも,それが保持されて溶出してくると,「一体何だろう?」と考え込んでしまいますね。この原因のひとつには,溶存空気量(UV検出の場合は特に溶存酸素量)の違いがあげられます。
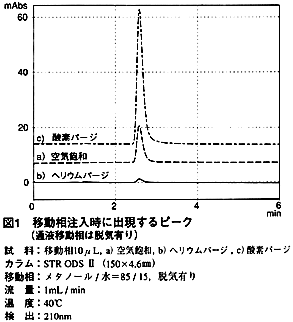
図1は,メタノール/水 混合液 移動相をオンライン脱気しているときに,その移動相を試料として注入したものです。溶存酸素濃度を変えて注入し,比較しました。試料(移動相)をそのまま,つまり空気飽和状態で注入すると,一つのピークが出現しています(図1a,10 μL注入時,約10mAU)。このピークは,試料(移動相)をヘリウムパージして(溶存酸素濃度はほぼゼロ)注入するとほとんど出なくなります(図1b)。また,酸素パージすると(溶存酸素濃度は空気飽和時の約5倍)大きくなります(図1c)。
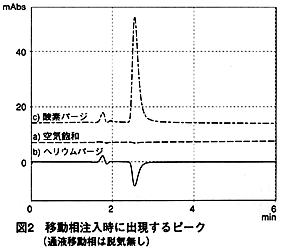
一方移動相がオンライン脱気されていない時には,試料(移動相)を空気飽和で注入してもピークはほとんど観察されません(図2a)。また,試料をヘリウムパージすればマイナスのピークが観察されます(図2b)。
これらの結果から,移動相と試料溶液中の溶存酸素量の違いが,ピークを出現させると考えられます。
2. ピークの大きさ
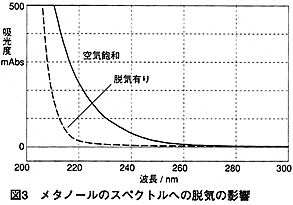
では次にそのピークの大きさを考えてみましょう。図3は空気飽和(脱気無し)のメタノールと,脱気したメタノールのスペクトル比較です。メタノールは脱気すると,吸収が小さくなります1) 。 吸収差は,波長によって異なり,210 nmでは300mAU以上,254 nmでは約10mAUになります。この値から,脱気しているメタノールに,空気飽和のメタノールを注入したときのピーク高さを単純計算します。流量1 mL/min,10 μL注入時に,仮にピークを三角形に見立てて底辺が0.4分間だとすると,ピーク高さは210 nmで15mAU以上,254 nmで約0.5mAUになります。短い波長では,結構大きいピークになりますね。
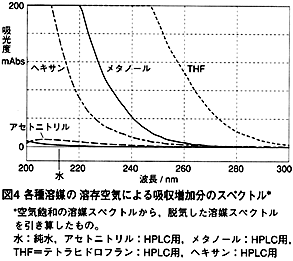
では他の溶媒はどうでしょうか?各種溶媒について,空気飽和の溶媒スペクトルから脱気した溶媒スペクトルを引き算した,差スペクトルを,図4に示しました。それぞれの溶媒の吸収は溶存空気により大きくなっていますが,水,アセトニトリルでは溶存空気の影響が小さく,ヘキサン,メタノール,THFでは影響が大きくなります。
さて,これらの吸収変化量と,溶媒の酸素溶解度との関係とは一致していません。例えば,ヘキサンはメタノールよりもはるかに酸素溶解度が大きい2) ですが,吸収変化は小さいのです。従って,吸収の由来は,酸素単独の吸収ではなく,酸素が溶媒と相互作用をしている結果と考えられます2) 。
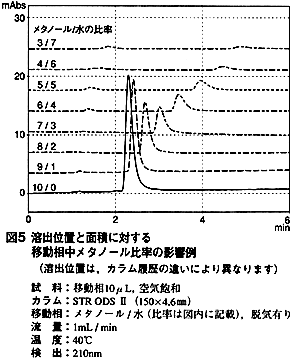
3. ピークの溶出位置
溶存酸素由来と考えられるピークは,早く溶出すると思われがちかも知れません。しかし,試料成分の保持挙動のように,有機溶媒比率を下げると溶出が遅れます。 図5はメタノール/水系の例で,移動相と試料溶媒は同じ溶媒組成です。このように,このピークは,目的成分と分離しない可能性があります。また,アセトニトリル/水系,あるいは水の代わりに緩衝液を使用しても,同様の傾向を示します。
なお,メタノール比率が下がるに従い,このピークは小さくなります。
4. 溶存空気由来らしきピークのチェック方法
気液分離膜脱気ユニットを使用中に,溶存空気由来なのか疑わしいピークがあった時には,次のようなチェック方法が考えられます。何れかに該当すれば,可能性が高いですね。
- 試料溶媒、注入量が同じ時には、ほぼ同じ溶出位置、大きさで出現している。 このとき試料溶液は、空気飽和にするため、撹拌(振り混ぜ)もしくは半開放系で放置する。 希釈して2倍容量注入すれば、約2倍の面積が得られる。
- 移動相(空気飽和)を注入して、現れるピークの溶出時間が、疑わしいピークの溶出時間と一致する。 このとき、移動相を脱気して(ヘリウムを10秒前後パージ)注入すると、ピークが小さくなる。
- 脱気ユニットを介さないで移動相を送液し、試料溶液を注入すると、ピークは小さくなる。 空気飽和の移動相を注入すれば、2.の時よりも必ず小さくなる。 試料溶媒組成が移動相と大きく異なれば必ずしも小さくなるとは限らない。
5. 対策
さて、溶存空気に由来するピークを完全に無くすことは困難ですが、小さくする方法を考えてみましょう(ここでは元条件がメタノール系移動相であるとします)。
- メタノール系移動相を、アセトニトリル(HPLC用)系に変更する。 変更に際しては溶出力や分離選択性 を考慮して行う 3) 。
→ 条件変更が許されるなら、この方法が良いです。 - 移動相中のメタノール比率を下げる。 このとき分析に適応出来るカラム(保持の弱いカラム、長さが短いカラム)を選択し直す。
→ ただし、ODSを放棄するのは汎用性に欠けますし、技術知識が必要です。 - 移動相のオンライン脱気をやめる。
→ やめると、流路中での気泡発生によるトラブルで定量精度が悪くなったり、検出の安定性が得られなくなる可能性があり1)、お勧めはできません。 - 試料溶液を注入前に脱気する。 ヘリウムで10秒前後脱気するだけで大きく脱気される。
→ ただし、作業が煩雑ですし、連続運転時にはあまり効果が期待出来ません。
また、対象成分との分離を改善する手だてとしては、
1) 目的成分がイオン性の場合は、移動相のpHを変えて目的成分の溶出位置を変化させる。
2) 有機溶媒の種類を変える。
などが考えられます。
これらは,基本的に分析メソッド開発時に検討すべきことになります。
以上のように,あるゴーストピークがどうやら溶存空気由来らしきことが判っても,容易には解決しない場合が多いかもしれません。それでも原因を把握しておくことは,分析条件を開発・管理される方々にとって,非常に大切ですね。
参考:
1) LCtalk特集号V「移動相の脱気」(1991).
2) S.R.Bakalyar, M.P.T.Bradley and R.Honganen, J.Chromatogr.,158, 277-293 (1978).
3) LCtalk vol.35, 8-9 (1995).
追伸:さて,同様の条件でもし検出器が屈折計の場合は,今度はマイナスピークを引き起こします。移動相はオンライン脱気されると,脱気されていないときよりも屈折率が大きくなるからです1)。このときは酸素だけではなく,窒素も関与します。こちらについても,機会をみて掲載します。(Y.Eg)