
お客様のご意見・ご要望のご紹介

田中 裕也 先生
東京工業大学 科学技術創生研究院 化学生命科学研究所 助教 (ご所属・役職は2020年4月発行時)
分子内を電子やホールといったキャリアが移動する現象は,有機電界効果トランジスタ(OFET)や有機電界発光素子(有機EL)などといった有機エレクトロニクスを始め,単一分子を分子素子として用いて電子回路構築を目的とする分子エレクトロニクスを実現する上で重要な基礎過程の一つである。混合原子価錯体は分子内に異なる原子価の金属種をもつ化合物群であり,1969年にCreutzとTaubeらによって発見されたルテニウム二核錯体(図1a)を端緒として広く研究されてきた1。その理由の一つとして,金属間の分子内電子移動挙動により電子移動速度や金属間相互作用を見積もることができ,これが結果として分子ワイヤー(分子導線)としての評価モデルの一つとして捉えられたためである。本稿では,混合原子価錯体における電子移動速度や電荷の非局在化の評価法の一つとして赤外分光法を用いる利点を述べた後で,最近の筆者らの例を紹介する。
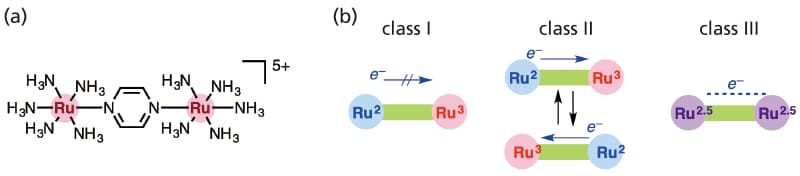
図 1 (a)Creutz-Taube 錯体,(b)混合原子価状態のRobin-Dayらの分類
混合原子価状態はRobin-Dayらの提案に基づき三つに分類することができる2。分子内にある金属種の原子価が全く異なり,その価数が区別可能なclass I,電子移動が十分早く,電荷が非局在化し複数の原子価数が平均化され全く区別のつかないclass III,その中間のclass IIである(図1b)。本分類は当初,固体無機化合物へ適用されていたものだが,混合原子価錯体においても同様に用いられて議論されている。実験的に分類する手法としては電気化学的手法や近赤外領域に見られる金属間に生じる電子遷移(IVCTバンド)をMarcus-Hush理論に基づき解析する方法が代表的である3, 4。その他にもESR(EPR)による電子移動速度の見積もり5 やMössbauer分光で金属の価数を直接判断する手法6などが用いられる。それぞれの手法で長所と短所があり,例えば電気化学的手法では用いる電解質や溶媒によってその結果が大きく影響されることや7,IVCTバンドの解析ではしばしば多数の吸収帯が見られて,その帰属が困難になることもある8, 9。またESRやMössbauer分光ではその測定のタイムスケールが電子移動速度よりも遅い場合があり,素早く電子移動する系では十分に評価できない。一方,赤外分光法(FT-IR)は分子の対称性・電子状態を解析できることから,混合原子価状態を評価するうえで非常に有用な手法となりうる。さらにその測定のタイムスケールが短く(10-12~10-13 s),早い電子移動系においても,その電子移動評価が可能となる。
著者らは金属アセチリドを主鎖骨格に持つ混合原子価錯体の合成とその物性評価を行っている。特に,分子ワイヤーを指向して電子豊富なd8遷移金属錯体のMCp*(dppe)(M = Fe,Ru,Cp*;pentamethylcyclopentadienyl,dppe;1,2-Bis(diphenylphosphino)ethane)を末端に有する一次元混合原子価錯体の合成とその展開を行ってきた10。最近では金属アセチリド分子ワイヤーの単分子伝導度計測まで展開している11, 12。一方,これら混合原子価錯体を二次元へ拡張した例は少なく,また一次元系に比べて,IVCT バンドが複数生じる可能性が高く,その解析は困難である8。一般にアセチレンのν(C≡C)伸縮振動は対称禁制のためFT-IR測定では非常に弱く観測されることが多いが,金属アセチリド系では十分強い強度で観測できる。そこで以下では,FT-IRを用いた二次元混合原子価金属アセチリド錯体の電子状態の評価について述べる。
ヘキサアリールベンゼンはベンゼン環の周囲に6つの芳香環を持つ化合物群であり,周辺芳香環は互いの立体反発を避けるために,中心ベンゼン環に対して傾いて配向している。その結果,周辺芳香環同士でπ─π相互作用が可能な配向をとるため環状の相互作用を示す13。これをトロイダル共役系もしくは環状共役系と呼び,この相互作用を活用した機能材料の開発が進められている(図2a)14。一方,周辺芳香環の構造物性相関については,ほとんど明らかにされていなかった。そこで,このような環状共役系を介した二次元混合原子価錯体の性質を調査した15。金属フラグメントとしてはFeCp*(dppe)を用い,周囲に六つのベンゼン環(1)およびチオフェン環(2)を有する六核鉄錯体を合成し(図2a),1,2のアセチレンを指標としたFT-IRによる混合原子価状態の評価を行った。まず電気化学的な性質をサイクリックボルタモグラムにて評価したところ,1と2共にピーク電位差が200 mV以上におよぶ非常に幅広い可逆な酸化波が1つ観測された。これは六つの鉄二価種が段階的に酸化して,六つの鉄三価種へ変化した過程を表しており,幅広い分布は金属間に複数の弱い相互作用が働いていることを示している。この酸化プロセス中に鉄二価と三価の混合原子価状態をとるが,各酸化波の電位差は非常に小さい。そのため溶液中では各酸化種が平衡混合物として存在するため,単離するのは困難であることが示唆された。
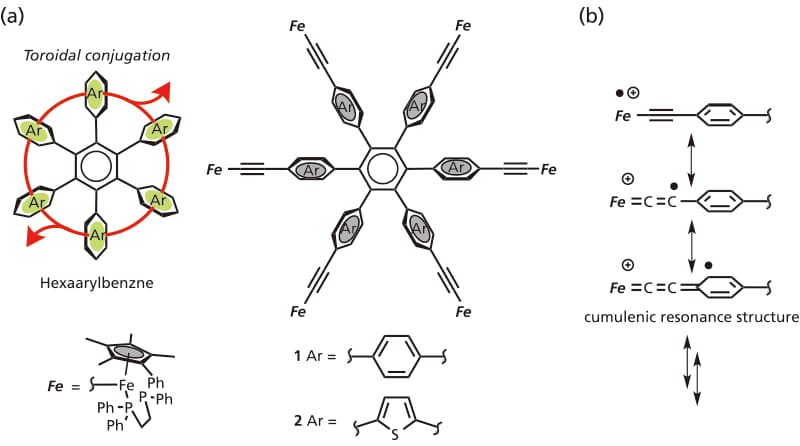
図 2 (a)1と2 の分子構造,(b)酸化種におけるクムレン共鳴構造
酸化種の電子状態を調べるために,ヘキサカチオン種[1]6+および[2]6+をフェロセニウムカチオンを酸化剤としてそれぞれ調製した。これと対応する中性種を任意の割合で混合した際のジクロロメタン溶液中でのFT-IR測定を行った(図3)。中性状態では1 は2053 cm-1,2 は2032 cm-1 にそれぞれν(C≡C)の非対称伸縮振動に対応するシグナルが観測された。金属からアセチレンへの逆供与結合の寄与により,有機アセチレン(2100~2200 cm-1)と比べて数十cm-1ほど低波数シフトしている 。一方,ヘキサカチオン種では[1]6+で1951,1989cm-1,[2]6+で1952 cm-1 にそれぞれν(C≡C)伸縮振動が観測された。中性種に比べて80-100 cm-1 程度低波数シフトしているが,これは酸化により金属上に生じたラジカルカチオンが架橋配位子側へ非局在化し,アセチレン構造からクムレン型の共鳴構造の寄与が増大したためであると考えられる(図2b)。興味深いことに1と2では混合した際のスペクトルに関して異なる挙動がみられた。中性種1とヘキサカチオン種[1]6+を5:1~1:5まで変化させると,その割合に応じて[1]n+(n = 1-5)が熱力学的に安定な比で生成するが,FT-IRスペクトルでは中性種1とヘキサカチオン種[1]6+で見られたν(C≡C)伸縮振動ピークと同位置に対応するピークが見られた。すなわち単純な足し合わせのスペクトルが観測されたことになる。一方,同様の操作を2と[2]6+ で行ったところ,[2]6+ の割合を増やしていくにつれピークのブロード化とシフトが認められ,単純な足し合わせとは異なるピークとなることが分かった。これは[2]n+(n = 1-5)の混合原子価状態に於いて,金属間での電子移動が生じて,金属種の原子価がIR のタイムスケールで二価と三価の中間的な値を取っていることを示している。すなわち[2]n+(n = 1-5)の中でclass II に分類される混合原子価状態をとるものが存在することが明らかとなった。一方ベンゼン誘導体1では電荷が完全に局在化しており,FT-IR においては金属間の相互作用は認められずclass Iもしくはサイクリックボルタモグラムの結果を考慮してclass I/II の境界領域に分類した。以上のようにFT-IRを用いることで,二次元骨格で架橋した混合原子価状態の電子移動に関する知見が得られる。
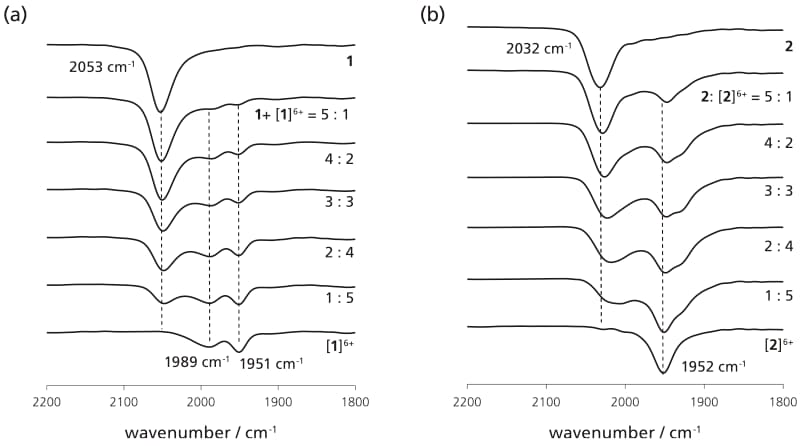
図 3 CH2Cl2 溶液中でのFT-IRスペクトル(a)1,[1]6+ およびそれらの混合状態,
(b)2,[2]6+ およびそれらの混合状態
続いてより強い相互作用を有する二次元混合原子価系を構築するために,ポルフィリンに着目した。これまでにポルフィリンのメソ位に酸化還元ユニットとして四つのフェロセンを導入した系がclass IIの混合原子価状態を示すことが報告されている17。そこで,酸化還元ユニットとしてメタルアセチリドを導入した四核ルテニウム錯体3を合成した(図4a)。電気化学的測定では,よく分離した四段階の可逆な酸化波が観測されたことから,各酸化状態が熱力学的に安定で単離が可能であることが示唆された。そこで,モノカチオン種とジカチオン種を別途合成し,単離を行った。FT-IR測定を行ったところ,中性体では2031 cm-1に見られたν(C≡C)伸縮に対応するピークが,モノカチオン種では1980 cm-1 に,ジカチオン種では1945cm-1 にそれぞれ低波数シフトして観測された。先の例とは異なり,各酸化状態で1本のν(C≡C)伸縮振動ピークが観測された。したがって金属間相互作用が十分大きく,電荷が全体に非局在化し,金属フラグメントの酸化数が等価なclass IIIであることが示された。
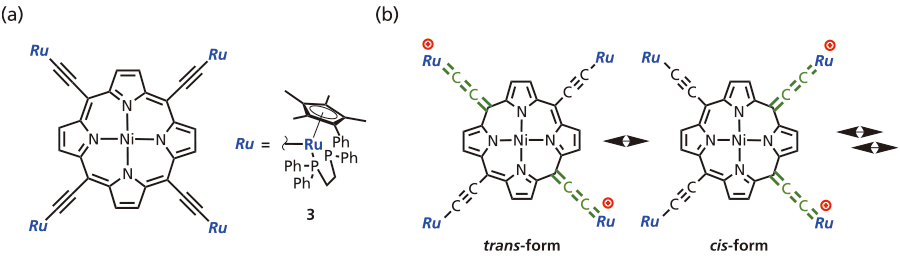
図 4 (a)3 の分子構造,(b)[3]2+ の共鳴構造の例
興味深いことにジカチオン種はNMR 活性・ESR 不活性であり分子内に不対電子が存在しないことが分かった。従ってアセチレンとクムレン構造を持つ共鳴構造体を考える必要がある(図4b)。しかしながらこの場合は,アセチレンν(C≡C)とクムレン構造ν(C=C)に由来する二つの伸縮振動が見られることになる。そのため以下二つのことが考えられる(1)異性体が熱障壁のある平衡状態にあり,異性化の速度が十分に早い,もしくは(2)共鳴状態にあり,平均化した基底状態をとる。もし(1)であれば温度降下により,異性化速度が遅くなり,各異性体を検出できる可能性がある。そこで低温下でのFT-IR測定を試みた。[3]2+ のKBr ペレットを調製し,室温から-180 ℃まで降温させた際のピークの変化を調査した(図5b)。その結果,若干の波数シフトは観測されたものの,アセチレンとクムレン構造に由来する伸縮振動は観測されなかった。従って現時点では(2)の基底状態が共鳴状態にあると考えている。これはジカチオン種の低温から室温までの温度可変結晶構造解析の結果からも支持された。このように,二つの異なる酸化状態で共にclass III に分類される化合物はこれまで報告になく,本系が非常に強い金属間相互作用を有することを示した18。
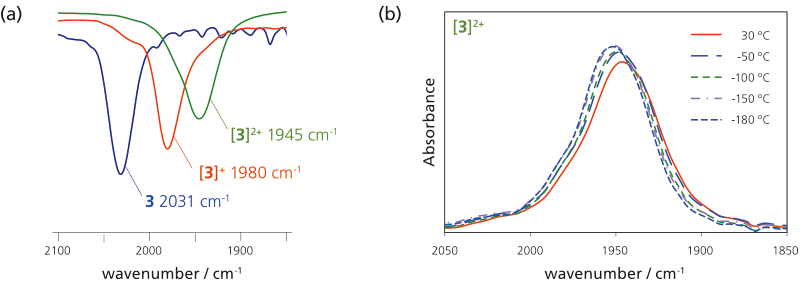
図 5 (a)[3]n+(n = 0-2)のCH2Cl2 溶液中でのFT-IRスペクトル,
(b)[3]2+ のKBr ペレット中での温度可変FT-IR
以上,本稿ではFT-IRを用いた二次元混合原子価錯体の電子状態評価について述べた。複雑な錯体系であっても適切にIR活性な官能基を導入することができれば,電子移動に関連する有用な情報を得ることができることがわかった。また密度汎関数法を用いた計算などを利用することで,実験値との比較をすることもでき,各種異性体の存在などをより詳細に議論できるが,本稿では紙面の都合により割愛させて頂いた。近年では有機合成化学の研究室であってもFT-IRを所有しないところも多いと聞くが,今後も有機・金属錯体材料の基礎物性を明らかにする上で,FT-IRが重要な役割を担うと期待している。
