
お客様のご意見・ご要望のご紹介

杉本 敏樹 先生(助教)
京都大学大学院理学研究科化学専攻 助教/国立研究開発法人科学技術振興機構 さきがけ兼任研究員 (ご所属・役職は2017年4月発行時)
完全水分解光触媒は水の酸化還元反応により水素と酸素を取り出す「光-化学エネルギー変換物質」として活発 に研究されている1,2)。光触媒の動作原理として,次のように大きく4つの素過程が存在することが知られている。①光誘起電荷(正孔と電子)の生成,②正孔・電子の分離と表面への拡散,③表面における正孔と電子の捕捉,④正孔による水の酸化反応,電子による還元反応。これらの素過程と共に,正孔と電子の再結合過程も存在する。再結合によって正孔と電子が消滅すると,酸化・還元反応を誘起する担い手が失なわれ,光触媒反応活性の低下につながる。
時間分解ポンプ・プローブ法を用いた光触媒の電荷ダイナミクスの研究から3-10),光誘起電荷が酸化・還元反応を誘起する(過程④)にはマイクロ秒以上の時間を要することが明らかにされている。したがって,電荷生成(過程①)の後フェムト秒からピコ秒のスケールで正孔と電子が分離し,表面へ拡散(過程②)した後は,これらの電荷は表面でマイクロ秒以上の時間スケールで再結合消滅を免れて存在し続けなければ酸化・還元反応(過程④)を誘起することができない。そのためには,正孔と電子が空間的に分離された状態で表面に効率的に捕捉(過程③)されなければならない。
著者らは近年,平均粒径5 nmのアナターゼ型二酸化チタン(TiO2)ナノ粒子(ST-01,石原産業)を10-2から103 Paの水蒸気雰囲気下に静置し,時間分解近紫外ポンプ・中赤外プローブ測定を行ってきた11)。その結果,図1に示すように,光誘起正孔の捕捉能力が水蒸気圧力100 Paまでは圧力と共に増大するが,100 Pa以上では圧力と共に減少することが明らかになった。一般に,水蒸気圧力が増大するにつれて表面吸着水の量は単調に増大する12-14)。したがって図1の結果は,100 Pa以上の圧力領域の吸着水が触媒表面あるいは吸着水層に何らかの構造・電子状態変化を誘起している事を示唆している。すなわち,吸着水が光触媒の物理化学的機能の発現に大きく関与していることを示唆している。本稿では,この吸着水の正体を明らかにするために行った一連の赤外振動分光の結果11)を紹介する。
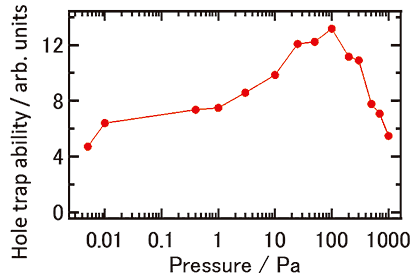
図1 TiO2ナノ粒子(ST-01)の正孔捕捉能の水蒸気圧力依存性11)。各圧力において、水分子は吸着脱離平衡状態にある。
飽和蒸気圧以下の圧力領域で室温の水蒸気の圧力を自在に制御するために15-17),10-3 Pa以上の絶対圧力を計測可能な隔膜真空計を備える高真空セルを作製した。このセルを拡散反射配置の光学系に組み込み,10-2 P aから103 Paの水蒸気雰囲気下で吸着脱離平衡状態にあるTiO2ナノ粒子(ST-01)表面吸着水の赤外振動分光を行った11)。
図2(a)に,赤外振動スペクトルの水蒸気圧力依存性を示す。1600 cm-1近傍に分子状吸着水の変角振動バンドが,3000 cm-1近傍に分子状吸着水あるいは解離吸着水(表面水酸基)による線幅が広い水素結合OH伸縮振動バンドが観測された。1×10-2 Paという高真空条件においてこれらのバンドが観測されるということは,水分子や水酸基がTiO2ナノ粒子表面に強く吸着している,あるいは粒子内部で水和物を形成している事を示唆する。これらのスペクトルが試料表面と内部のどちらに存在する水分子・水酸基に由来するのかを確かめるために,試料を2000 Paの重水(D2O)蒸気に曝して赤外振動スペクトルを測定した(図2(b))。その結果,OH伸縮振動バンドとHOH変角振動バンドが消失し,2300 cm-1近傍と1200 cm-1近傍にOD伸縮振動バンドとDOD変角振動バンドが現れた。これらの結果から,1×10-2 Paで観測されているスペクトルが試料表面に存在する分子状吸着水と水酸基に由来することが明らかになった。ちなみに,700 Kまで昇温させた際の脱離挙動(スペクトルの面積を吸着量とみなした吸着等圧線)の解析から,これらの分子状吸着水,あるいは表面水酸基の吸着エネルギーは100 kJ/molから200 kJ/molにかけて分布していることを確かめている11)。
変角振動バンド,及び水素結合OH伸縮振動バンドの面積の水蒸気圧力依存性を図2(c)に示す。いずれのバンドも,圧力が増大するにつれて面積が増大している。これらの面積は水蒸気圧力の減少に伴って可逆に減少することが確かめられた。一般に,水酸基は表面に強く吸着して室温では可逆に脱離しないため,これらの結果は,水蒸気圧力の増加に伴って非解離の水分子が吸着していることを示唆している。ここで,水素結合OH伸縮振動バンドと変角振動バンドの面積の増加曲線は100 Pa以下では吸着量(分子層(ML)単位)の増加曲線と重なっているが,100 Pa以上では水素結合OH伸縮振動バンドの増加曲線が吸着量よりも大きくなっている。OH伸縮振動の振動子強度は周囲との水素結合環境に大きく依存し,水素結合する水分子の配位数が大きくなるにつれて振動子強度は増大する18,19)。したがって,OH伸縮振動バンドの面積の増加曲線は,100 Paの水蒸気圧力前後で表面吸着水の水素結合状態(配位状態)が変化している事を示唆している。一方,変角振動の振動子強度は周囲の環境にほとんど依存しない20-22)。したがって,変角振動バンドの面積は吸着量にほぼ比例して増大する。
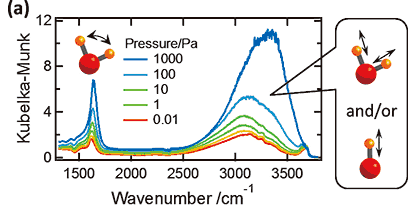
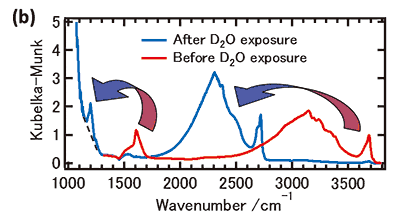
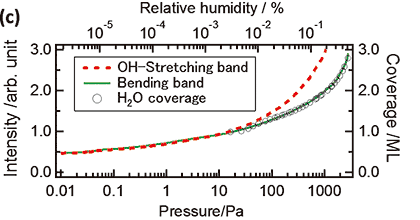
図2 (a) 代表的な水蒸気圧力における,ST-01表面吸着水の赤外振動スペクトル11)。(b) 0.01 PaのH2Oガス雰囲気下,及び2000 PaのD2Oガス雰囲気に曝露した後に約0.01 PaのD2Oガス雰囲気下で測定した赤外振動スペクトル11)。(c)水分子の吸着量(白丸),水素結合OH伸縮振動バンドの強度(赤破線),及び変角振動バンドの強度(緑実線)の圧力依存性。点線と実線は10-2から10-1Paにおける値が一致するようにスケーリングしている。白丸に示す吸着量の圧力依存性は,20から80 Paの値が実線と一致するようにスケーリングしている。
本節では水素結合OH伸縮振動バンドのピーク波数やスペクトル形状に焦点を当て,水蒸気圧力100 Pa前後で吸着水の水素結合ネットワークがどのように変化しているのかを議論する。水蒸気圧力が増加するにつれてOH伸縮振動は振動子強度が増加し(図2(c)),そのスペクトルは高波数側にブルーシフトしている(図2(a))。103 Pa以上の圧力領域のスペクトル形状は,液体水のスペクトル形状23)と酷似している。したがって,103 Pa以上の水蒸気雰囲気では試料表面に液体水と類似した水素結合ネットワーク構造を持つ吸着水が凝集していると考えられる。以下では,スペクトル形状の圧力依存性に焦点を当てて議論を行う。
ピーク強度で規格化した差スペクトル(I(P+ΔP)-I(P))/I max(P)の水蒸気圧力依存性11)を図3(a)に示す。差スペクトルの形状は,10-2から10 Paの圧力領域では有意に変化していないが,10から103 Pa にかけて線幅が減少しながらブルーシフトし,103 Pa以上の圧力領域では再び有意な変化を示さなくなっている。図3(b)に,10-2から10 Paの圧力領域の差スペクトル形状(Aピーク,ピーク波数~3100cm-1),及び103 Pa以上の圧力領域の差スペクトル形状(Cピーク,ピーク波数~3400 cm-1)を示す。これらの2ピークに形状未知の1ピーク(Bピーク)を加え,全圧力領域のOH伸縮振動スペクトルに対して3ピークの線形結合でグローバルフィッティングを行った。グローバル解析から求められたBピークの形状を図3(b)に示す。10から103 Paで観られるブルーシフト(図3(a))は,主にこの3300 cm-1にピークを持つB成分の増加によって説明する事ができる。直観的には,Aピークは表面第1層に吸着した水分子のスペクトル,Bピークは表面第2層に吸着した水分子のスペクトル,液体水のスペクトルに類似したCピークは表面第3層以上の多層吸着水のスペクトルと考えることができる。
図3(c)に,グローバル解析における3成分の線形和の係数から求めた,各成分の寄与の水蒸気圧力依存性を示す。
ただし,ここではOH伸縮振動バンドに対応する変角振動バンドについてのグローバル解析で得られた各成分の線形和の係数を,図2(c)の対応関係から吸着量に変換して縦軸としている。100 Pa以下のA成分の増加の圧力依存性は2種類の吸着サイトを仮定したLangmuir吸着等温式でよく再現でき,その結果から,圧力増加と共に形成される第1層吸着水の吸着エネルギーは71 kJ/molと60 kJ/molと見積もられた11)。また,100 Pa以上のA,B,C成分の和の圧力依存性はBET吸着等温式でよく再現でき,その結果から第2層吸着水,及び多層吸着水の吸着エネルギーはそれぞれ52 kJ/mol,46 kJ/molと見積もられた11)。後者の値は,水の凝集エンタルピーと良く一致している。
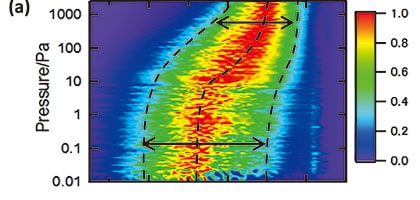
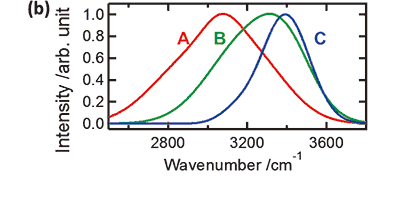
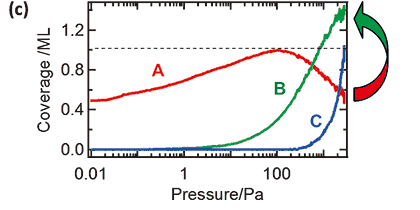
図3 (a) ピーク強度で規格化した差スペクトル(I(P+ΔP)-I(P))/I max(P)の水蒸気圧力依存性11)。
横軸は波数,縦軸は水蒸気圧力の二次元プロット。
(b) グローバル解析で用いられた3成分のスペクトル,及び(c) 3成分の寄与の圧力依存性11)。
図3に示す結果は,次のような点で興味深い。第2層吸着水の量が0.4 ML以上になる100 Pa以上の圧力領域において,第1層吸着水に由来するA成分の寄与が減少に転じ,その減少分だけ余分にB成分の寄与が増加している。本節ではA成分のレッドシフトの起源を考察し,100 Pa以上の圧力領域でみられるA→B成分転化の起源を議論する。
OH伸縮振動の振動数は,その分子が感じている局所的な水素結合環境に大きく依存して変化する18-20)。図4に,OH伸縮振動波数の局所電場強度依存性18)を示す。水素結合に全く関与しないOH振動子の場合はOH振動子方向の実効電場は0.1 V/Åオーダーであり,その振動波数は3700 cm-1程度である。ところが,周囲の水分子との水素結合やイオンとの化学結合によって水分子の電子雲が大きく歪まされると,OH振動子が感じる実効的な局所電場は増大する。4 V/Åの局所電場を感じる環境下ではOH振動波数は2800 cm-1程度となり,実に900 cm-1もレッドシフトする(図4)。
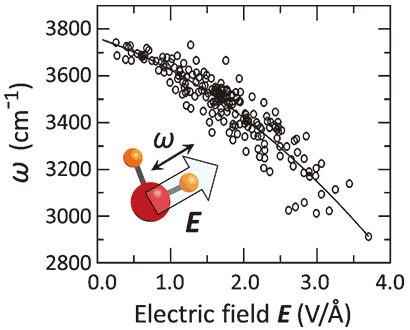
図4 水分子のOH伸縮振動波数と,
OH振動子方向の局所電場強度の相関18)。
では,TiO2表面上の吸着水は表面とどのように相互作用しているのだろうか? 第一原理分子動力学(MD)計算24)によって,表面に存在する(酸素イオンが)5配位の低配位Tiカチオンは,バルクの6配位Tiカチオンに比べて電子が過剰に存在する状態にあり,Lewis塩基として吸着水分子に対して部分的に電子供与することが報告されている。
この部分電子供与により吸着水分子は僅かに負に帯電し,それと共に電子雲の分布が変化する。さらにこの吸着水のOHがTiO2表面の酸素イオンや隣接する吸着水分子の酸素原子と水素結合を形成すると,このOH周辺の電子雲の分布はさらに変化し局所電場強度が増大する24)。その結果,低配位Tiカチオンに吸着した水分子の水素結合OH振動子の振動数は著しいレッドシフトを示しうる。
図3(b)に示すように,A成分のスペクトルは顕著にレッドシフトしている。本研究で用いたST-01ナノ粒子は球形に近い形状を有し,その粒子直径は約5 nmである11)。TiO2の球状ナノ粒子の表面には,平坦なテラス表面と異なり,酸素イオンが4配位の低配位Tiカチオンが存在している25)。
したがって,これらの4配位Tiカチオンが強いLewis 塩基点として吸着水分子に電子供与し,その結果として,第1層吸着水に起因するA成分は顕著なレッドシフトを示していると考えられる。それに対して,B成分の水分子は表面の低配位Tiカチオンとの直接的な相互作用が弱くなった結果として,A成分ほどのレッドシフトを示さなくなっていると考えられる。したがって,第2層吸着水が0.4 ML程度凝集し始める100 Pa程度からA成分の一部がB成分に転化するという図3(c)の結果は,図5に示すように,第2層吸着水との水素結合形成によって第1層吸着水の一部が低配位Tiカチオンとの結合を(部分的に)切断していることを示唆する。
ST-01は大きな曲率を有する球状ナノ粒子であるため,低配位Tiカチオンとの相互作用を優先して第1層が吸着すると,その水素結合ネットワーク構造はバルク水の本来の水素結合ネットワーク構造よりも大きな構造歪みを内包していると考えられる。100 Pa以上では,第2層吸着水との水素結合形成に伴ってこの構造歪みを解消する構造緩和が誘起され,その結果として低配位Tiカチオンとの結合が切断されていると考えられる。水分子の多層吸着に伴って表面第1層の水分子吸着層に構造緩和が誘起されるという現象は,第一原理MD計算によっても報告されている26)。
水蒸気圧力100 Pa以上の圧力領域でみられる第1層吸着水の構造変化は,同圧力領域で光誘起正孔の捕捉能が減少に転じるという図1の奇異な結果を合理的に説明することができる。非常にレッドシフトしたAピークのスペクトルを示す第1層吸着水は表面の低配位Tiカチオンからの部分電子供与により負に帯電している24)。したがって,この水分子は,正の電荷を有する光誘起正孔を静電的に引き込み有効に捕捉可能であると考えられる(図5)。それに対し,低配位Tiカチオンとの結合を切断してB成分のスペクトルを示すように構造緩和してしまった第1層水分子は,低配位Tiカチオンからの電子供与を受けらないので電気的には中性に戻ってしまっている。したがって,B成分に転化した第1層吸着水は光誘起正孔を静電的に引き込むことが困難となり,その結果として表面第1層吸着水の正孔捕捉能力が失なわれてしまっていると解釈できる(図5)。実際に,正孔捕捉能とA成分の寄与の水蒸気圧力依存性は非常によく相関している(図1,図3(c))。
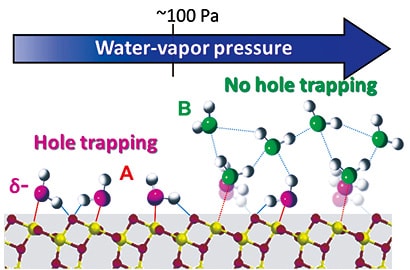
図5 TiO2ナノ粒子表面における吸着水の構造変化の模式図。
本稿で紹介してきたように,球状TiO2ナノ粒子(ST-01)の表面吸着水に対して赤外振動分光を行うことで,正孔捕捉能が吸着水の凝集によって非単調に増減するという現象(図1)に物理化学的解釈を与える事ができた11)。正孔は水分解反応の律速過程27)である酸化反応を誘起するため,本成果は,反応活性を増大させるための光触媒開発の表面エンジニアリングの指針と成り得る。
線幅が非常に広い水素結合OH伸縮振動バンドの一次元スペクトルから水分子の物理化学的機能に関する分光情報を有効に抽出することは一般に困難であり,また多くの場合そう信じられている。水素結合OH伸縮振動スペクトル測定結果自体はこれまで無数に報告されているが,物理化学的に有用な分光情報が抽出された例は数少ない。本稿で紹介してきたスペクトルの測定・解析により,水素結合OH伸縮振動バンドの分光学的研究の価値が再認識されると幸いである。また,二次元相関分光法23,28,29)を用いた発展的な分光研究が精力的に行われることも期待したい。
本稿で紹介した研究結果は,京都大学大学院理学研究科化学専攻の白井健次,渡邊一也,松本吉泰,治田充貴,倉田博基,各氏との共同研究や有益な議論に依っており,ここに感謝申し上げます。
