
お客様のご意見・ご要望のご紹介

鎌田 慶吾 先生
東京工業大学 科学技術創成研究院 フロンティア材料研究所 准教授 (ご所属・役職は2021年9月発行時)
複合酸化物はその構造に由来した電子的および磁気的性質に基づき,半導体・圧電体・電子材料・磁性材料など幅広い分野へと応用されている重要な無機化合物のひとつである。触媒の分野においても,近年の合成・測定技術だけでなく理論計算等の進歩により,その酸塩基あるいは酸化還元能に基づいた優れた触媒作用について多くの報告がなされている。一方,固体である複合酸化物は分子である均一系金属錯体触媒や有機触媒と比べると,望みの反応を達成するための触媒設計や精密合成に今なお多くの課題を抱えている。また,基質や反応剤などの分子活性化や構造 - 反応性の相関などの固体触媒上での詳細な反応機構に関する知見についても不明瞭な点が多く,さらなる高機能触媒の設計・合成を困難なものとしている。
このような研究背景の下,我々の研究グループでは,複合酸化物でありながらアニオン性クラスター分子である「ポリオキソメタレート」を基盤とした高機能触媒の設計に対する新しいコンセプトの立案・方法論の開拓と,それらを用いた環境調和型な実用的触媒反応系の開発に関する研究を展開した。[1] また,これらポリオキソメタレート分子に関する触媒研究で得た知見や概念を生かした新しい固体触媒の開発にも着手し,分子状酸素(O2)のみを酸化剤とした酸化触媒として機能する「ペロブスカイト酸化物」や二元機能酸・塩基触媒として機能する「金属ホスフェート」を開発している(図1)。[2]‒[9] 本稿では,これら2つの固体材料による酸化および酸塩基触媒作用に関連して,FTIRを用いた分子活性化に関する機構解明について紹介する。
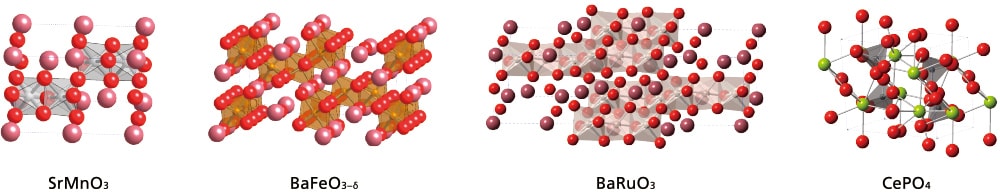
図1 ペロブスカイト酸化物(SrMnO3, BaFeO3‒δ, BaRuO3)と金属ホスフェート(CePO4)の結晶構造
選択酸化反応は全化学プロセスの3割を占め,多様な天然炭素資源を有用な酸化生成物に変換する化学工業的にも重要な反応の一つである。酸化剤として分子状酸素(O2)のみを用いるメリットにも関わらずO2酸化の有用性や適用性は未だ限定的であり,特にO2を温和な条件下で活性化でき広範な基質に適用できる不均一系酸化触媒の報告例はごく僅かである。我々は,錯体重合法を用いて合成した六方晶SrMnO3ペロブスカイト触媒がO2を用いた種々の基質の選択酸化反応に対して,優れた不均一系触媒として機能することを見いだした。[7] O2を用いた種々のMn化合物による1-フェニルエタノールの酸化反応に対する触媒効果を検討したところ,SrMnO3が最も高いアセトフェノン収率(83%)を示し,SrMnO3(25 m2 g-1)よりも大きな表面積をもつ活性化MnO2(122 m2 g-1)の収率(59%)よりも高かった。他のMn2+やMn3+を含むMn酸化物や錯体は本反応条件では不活性であった(1-6% 収率)。リーチングテストより反応は固体表面で進行していることが示唆され,ろ過により回収した触媒は活性の低下なく再使用可能であった。SrMnO3触媒はO2を用いた2級および1級のベンジル・アリル型アルコールの酸化反応に適用可能であり,対応するカルボニル化合物を与えた。
SrMnO3触媒によるアルコール酸化の反応機構について検討した(図2)。格子酸素が反応に寄与するMnO2ではO2・Ar雰囲気下においても反応が進行するのに対して、SrMnO3はO2存在下でのみ酸化反応が大きく促進されSrMnO3触媒によるO2活性化機構が示唆された(図2(a))。そこで表面酸素種についてO2吸着IRスペクトルの測定を検討した(図2(b))300℃で真空加熱処理を行ったSrMnO3に16O2を室温で導入したところ、金属スーパーオキソ種のv(O-O)(1200-1100cm-1)と同程度の位置である1152 cm-1に新しい吸収帯が観測された。この吸収帯はMnO2では観測されなかった。この吸収帯は真空排気により消失した。同様の条件下において18O2を吸着させたSrMnO3のIRスペクトルでは1086 cm-1に吸収帯が観測され、その同位体シフト値は理論値と一致した。Mnスーパーオキソ錯体の単離やLa1-xSrxMnO3上でのO2還元に関する理論的検討は報告されているが、SrMnO3上でのO2還元的活性化とその触媒応用の報告例はこれまでにない。上記FTIRによる検討と速度論分析から、可逆的に生成したMnスーパーオキソ種が本酸化反応において重要な役割を果たしていると推測した(図2(c))
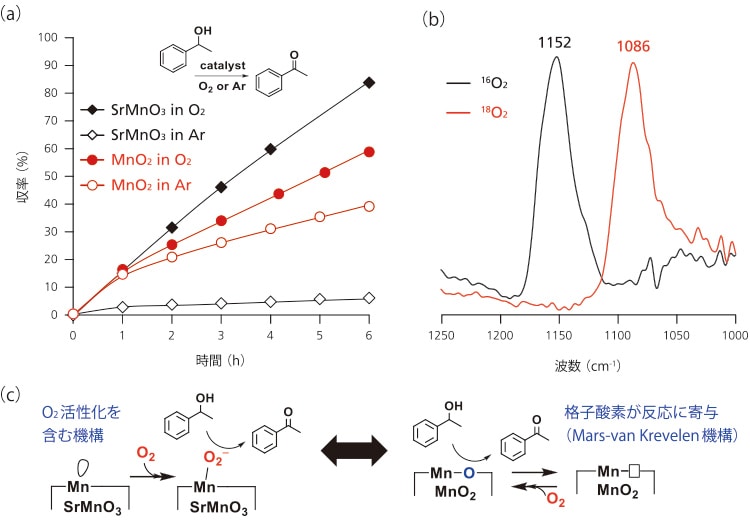
図2 (a)Mn酸化物によるO2を用いた1-フェニルエタノール酸化反応の経時変化
(b)16O2および18O2を吸着させたSrMnO3のIRスペクトル
(c)Mn酸化物によるアルコール酸化の推定反応機構
また,多段合成と元素適用性に課題を抱える錯体重合法に代わり,ペロブスカイトナノ粒子合成時のアモルファス前駆体生成の重要性に着目した「アミノ酸(アスパラギン酸)を用いた単純かつ効率的な新合成ルート」を開発した。[2]-[6] アスパラギン酸と金属酢酸塩を用いて調製したアモルファス前駆体の低温焼成により高表面積(~50 m2 g-1)な六方晶SrMnO3の合成に成功し,錯体重合法で合成したサンプルよりも高い触媒性能を示すことを見いだした。この合成手法は様々な元素の組み合わせに適用することができ,高原子価鉄を含む六方晶6H-BaFeO3-δや菱面体晶BaRuO3などのナノサイズのペロブスカイト酸化物合成が可能であった。六方晶6H-BaFeO3-δが常圧O2のみを酸化剤としたアダマンタンを含む種々のアルカン類の酸化反応やアルケン類の酸化的C=C 切断反応において効率的かつ再使用可能な固体触媒として機能した。[3][4] この研究が添加剤などを使用することなくO2のみを用いたアダマンタンの酸化反応に対して自然界に多く存在する鉄酸化物ベースの初めての固体触媒の報告例である。また,菱面体晶BaRuO3ナノペロブスカイトが,O2のみを酸化剤とした種々の芳香族および脂肪族スルフィドの選択酸化反応に対して優れた不均一系触媒として機能した。[5]
酸 - 塩基能をもつ複数の触媒活性点による協奏的な分子活性化は,特異的な触媒活性や選択性の発現に寄与することが知られている。不均一系触媒の分野においては,均質かつ構造制御された酸塩基活性点の構築が困難であるため,触媒構造のファインチューニングや反応性の制限といった点において課題を抱えている。したがって,新しい高活性な酸・塩基無機固体触媒の設計と開発は重要な研究課題の一つである。このような研究背景の下,我々はLewis酸触媒として機能する希土類金属と特異的塩基触媒作用を示すオキソアニオンの複合化により得られる希土類リン酸塩が,求核剤と求電子剤の両方に作用し優れた酸塩基二元機能触媒として機能するのではという着想に至った。本項では水熱法により合成した単斜晶CePO4触媒によるカルボニル化合物の高官能基選択的アセタール化反応とFTIRによるプローブ分子を用いた酸塩基触媒能について紹介する。[8]
単斜晶CePO4は,Ce(NO3)3と(NH4)2HPO4を水熱条件で反応させることで合成した。CePO4の走査電子顕微鏡(SEM)測定から,ナノロッド状粒子であることが明らかとなった。種々の酸塩基触媒の存在下,メタノールを用いた水酸基とアルデヒド基を有する糖由来化合物の5-ヒドロキシメチルフルフラール(HMF)のアセタール化反応を行った。種々の触媒の中でも,CePO4がHMFのアセタール化反応に最も高い活性を示しアセタール体を78%収率で与え,アセタール選択率は96%に達した。触媒が存在しない場合には反応は進行せず,均一系酸あるいは塩基触媒自体はHMFの官能基選択的アセタール化反応に対して不活性であった。酸塩基触媒として機能することが報告されている金属酸化物について検討した。Nb2O5はCePO4よりも本アセタール化反応に有効ではなく,他の金属酸化物(SiO2,ZrO2,CeO2,Al2O3,MgO,TiO2,SnO2)はほぼ不活性であった。典型的な固体酸触媒は生成物の複雑な混合物を与え,触媒前駆体の混合物は本アセタール化反応に有効ではなかった。 CePO4存在下,種々のカルボニル化合物とアルコールとの組み合わせから対応するアセタール化合物へと高収率で変換可能であった(図3)。本触媒系はグラムスケールでのメタノールを用いたHMF のアセタール化やグリセロールを用いたアセトンの位置選択的アセタール化反応に適用可能であった。
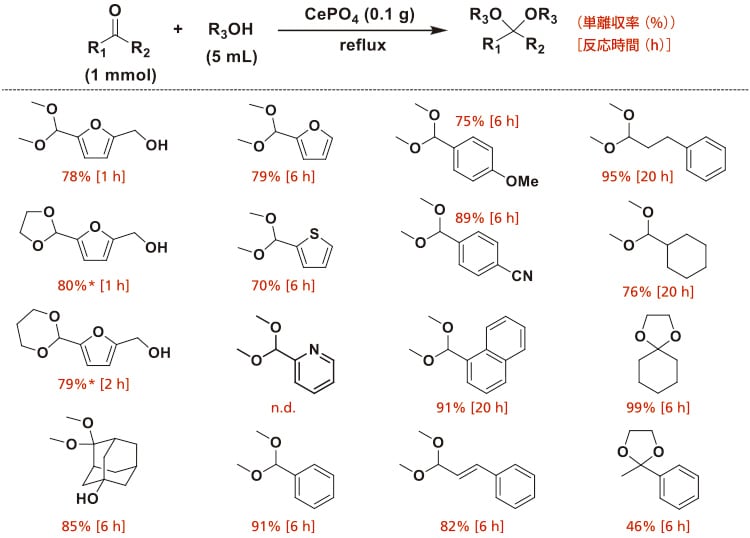
図3 CePO4触媒によるアセタール化反応の基質適用性
ピリジンをプローブ分子とした吸着IR測定によりCePO4の酸性質を評価した(図4(a))。ピリジンを吸着させたCePO4のIRスペクトルから,Lewis酸点に配位したピリジン種に帰属される1445 cm-1の吸収帯が観測され,Brønsted酸点に結合したピリジニウムイオン由来の1540 cm-1の吸収帯は観測されなかった。吸収帯強度から見積もったCePO4上のルイス酸量は0.096 mmol g-1であった。クロロホルムを吸着させたCePO4のIRスペクトルでは,クロロホルム分子のv(C-H)が3034 cm-1から3008 cm-1に低波数シフトしたことから,表面塩基点の存在が示唆された(図4(b))。さらに,酸点と塩基点に同時相互作用することに由来するクロロホルム分子のδ(Cl-C-H)に帰属される1250 cm-1の新しい吸収帯が現れた。したがって,CePO4上の塩基点はルイス酸点近傍に存在し結晶構造と良い一致を示した。
CeO2も優れた酸塩基触媒として機能することが知られているにも関わらずCePO4のみがアセタール化反応に高い触媒活性を示したことから,CePO4とCeO2による基質の活性化モードをアセトンおよびメタノール吸着させたサンプルのIR測定により検討した。CePO4上に吸着したアセトンのv(C=O)に帰属される強い吸収帯(1699 cm-1)は気相のアセトン(1731 cm-1)よりも低波数に観測された(図4(c))。一方,CeO2上に吸着したアセトンのIRスペクトルでは,異なる酸点に配位したアセトン分子(1700 cm-1と1673 cm-1)とアルドール縮合生成物に帰属される吸収帯(1627 cm-1と1570-1550 cm-1)が確認された(図4(d))。これは,アルドール縮合反応を促進することなくCePO4上の均質なLewis酸点がケトンのカルボニル酸素と相互作用していることを示唆している。
メタノールを吸着させたCePO4のIRスペクトルでは,v(O-H)領域で3000-3500 cm-1のブロードな吸収帯と負のOH吸収が観測されると共に,2952と2849 cm-1とにそれぞれva(s CH3)とv(s CH3)に帰属される吸収帯が観測された(図4(e))。このようなブロードな吸収帯とv(CH3)の吸収帯位置から,メタノールが水素結合を介してCePO4上に分子状で吸着していることが示唆された。一方,CeO2上に吸着したメタノールのIRスペクトルでは2911と2805 cm-1にそれぞれメトキシド種のvas(CH3)とvs(CH3)に帰属される吸収帯が観測された(図4(f))。したがって,CePO4は均質なLewis酸点と弱い塩基点がそれぞれHMFとメタノールと相互作用する二元機能触媒として機能することで,高効率かつ官能基選択的なアセタール化反応を達成したと考えられる。
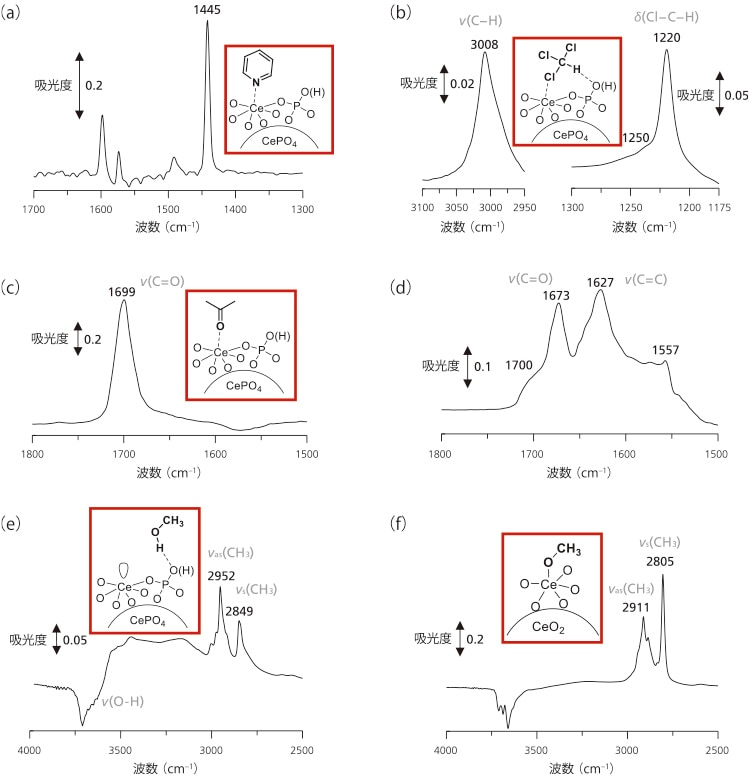
図4 (a)ピリジン,(b)クロロホルム,(c)アセトン,
(e)メタノールを吸着させたCePO4のIRスペクトル,
(d)アセトン,(f)メタノールを吸着させたCeO2のIRスペクトル
本研究では,結晶性複合酸化物の特異的構造に着目することで,O2酸化反応に有効な面共有複核サイトをもつ六方晶ペロブスカイト型酸化物や均質な酸塩基点をもつ二元機能金属ホスフェートを開発した。また,構造と機能の相関解明において,FTIRを用いた固体表面上での分子活性化機構の検討が極めて有効であることも確認できた。これらの知見は,酸素欠陥形成エネルギーの低い酸素原子をもつβ-MnO2などの固体材料のナノ構造制御や反応開発にも生かされている。[10]-[13] 今後は,上記のような結晶性金属酸化物の更なる高機能化(微粒子化,形態制御,配向制御など)による高難度反応の実現や,計算化学的アプローチ(マテリアルインフォマティックスなど)を利用した新材料に基づくブレイクスルーの創出,という重要課題を実現するための金属酸化物触媒の開発が期待される。また,我々の開発したナノ材料は熱以外の電場や電気化学エネルギーを用いた触媒反応系への応用も可能である。[9], [14]-[16]今後は,実験化学を基盤として結晶性金属酸化物の更なる高機能化(微粒子化・形態制御・配向制御など)による高難度反応の実現を目指すと共に,理論計算との融合による新物質に基づくブレイクスルーの創出に取り組んでいきたいと考えている。
上記の研究内容は,東京工業大学科学技術創成研究院フロンティア材料研究所(原亨和教授研究室)の研究設備を利用して得られた成果である。また,第一原理計算は同研究所の大場史康教授・熊谷悠准教授に行っていただいた。上記先生方をはじめとする研究室構成員皆様および共同研究者の先生方(早稲田大学関根泰教授・小河脩平准教授,東京工業大学山口猛央教授・菅原勇貴助教)のご指導,ご協力に深く感謝いたします。
