ゲノム編集による変異導入の確認
新着情報
■ ゲノム編集用途でのユーザーレビューを掲載(下記バナーからご覧ください)



ゲノム編集による変異導入の確認
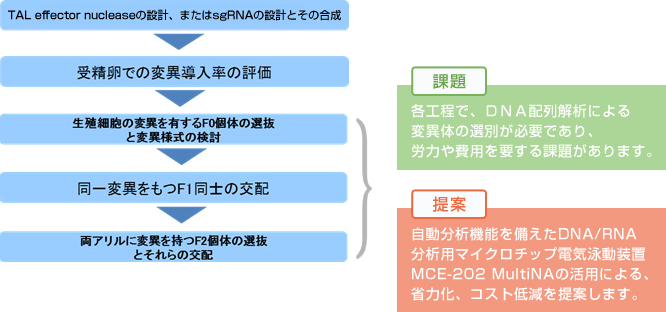
ゲノム編集を用いた変異系統を確立するためには,様々な工程で変異体を選別する必要がありますが,変異導入の確認には費用や労力を要することが課題として挙げられます。ここでは、DNA/RNA分析用マイクロチップ電気泳動装置 MCE-202 MultiNAを用いたヘテロ二本鎖移動度分析(HMA:Heteroduplex Mobility Assay)による効率的な変異導入の確認について紹介します。

-
ゲノム編集とは
-
変異導入と変異系統作製までの流れ
-
変異導入の確認: 「ヘテロ二本鎖移動度分析とMultiNAの活用」
-
■ヘテロ二本鎖移動度分析
-
■DNA/RNA分析用マイクロチップ電気泳動装置 MCE-202 MultiNA
-
■ヘテロ二本鎖移動度分析による変異導入した個体の選別例
各項目をクリックすると詳細な説明が表示されます。
ゲノム編集とは
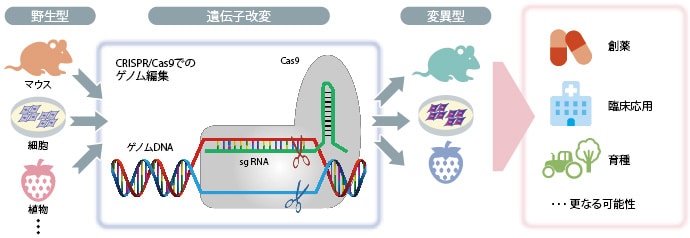
ゲノム編集とは,ゲノム上の特定部位を切断できるように設計された人工制限酵素(人工ヌクレアーゼ)を用いて,ゲノム配列に任意の欠失,挿入,置換といった変異導入を行う遺伝子改変技術です。 これまでZFN(zinc-finger nuclease),TAL effector nuclease(Transcription activator-like effector nuclease),そしてCRISPR/Cas9(Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR associated protein)といった人工制限酵素がゲノム編集ツールとして利用されており,特に第3世代であるCRISPR/Cas9は標的配列の設計が容易なことから急速に普及しています。
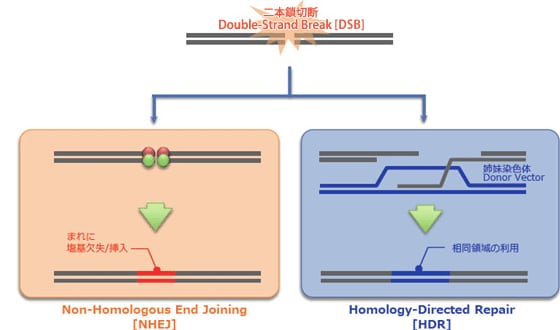
これらの人工制限酵素を発現するベクター,もしくは人工制限酵素自体がマイクロインジェクション法やエレクトロポレーション法などで細胞内に導入されると 標的配列でゲノムが切断されますが,細胞は主に2つの経路でこの切断を修復します。その1つは,細胞周期に依存しない非相同末端結合(NHEJ:Non- Homologous End Joining)です。 この経路ではゲノム切断という急場をしのぐため“ とりあえず” 切断を受けたゲノムを直接つなぎますが,その過程において微小な塩基の欠失や挿入が発生することがあります。このランダムに発生する変異導入を利用することで,意図的にフレームシフトを誘発し,遺伝子を破壊(ノックアウト)することができます。
一方,もう一つの経路は正常な姉妹染色体を鋳型として利用する相同組み換え修復(HDR:Homology-Directed Repair)です。 この経路では,ゲノムの切断が元通りに修復されるため非相同末端結合に見られるような変異は導入されません。しかしながら,相同性 をもつ2本鎖DNA (または1本鎖DNA)をドナーとして細胞内に導入することで,ゲノム内にドナー由来の配列が取り込まれるため遺伝子挿入(ノックイン)や遺伝子改変が可能となります。
このようなゲノム編集を用いた遺伝子改変法は,自然偶発的な組み換えを拠りどころとしていた従来法に比べ,飛躍的に改変効率を向上させました。そして,これまではマウスなどに代表されるES細胞が利用できる動物のみに遺伝子改変が可能でしたが,魚類,両生類,爬虫類や大型哺乳動物等といった従来は作製が困難であった動物に対する遺伝子改変が可能となりました。その他にも,昆虫や植物等に対する遺伝子改変にゲノム編集は利用されています。ゲノム編集の応用範囲は基礎研究に留まらず,ヒト遺伝子疾患モデルの作製や遺伝子治療への展開を目的とする医療分野に加え,動植物の品種改良といった分野で大きな期待が寄せられています。
変異導入の確認:「ヘテロ二本鎖移動度分析とMultiNAの活用」
■ ヘテロ二本鎖移動度分析
遺伝子ノックアウトを目的とした非相同末端結合での変異導入では,1塩基~数十塩基 の微小な欠失や挿入が生じます。 この変異を確認するためには変異導入部のDNA塩基配列解析が不可欠となりますが,一般的にDNA塩基配列解析は労力・ 費用を要するため,先に検体を選別した方が効率的です。その選別方法として,変異近傍領域のPCRと電気泳動で実施できるヘテロ二本鎖移動度分析は,簡便 かつ有用な方法として利用されてきました。
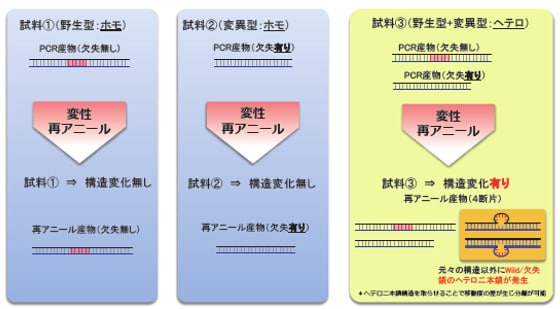
同じ長さの二本鎖DNAを電気泳動する際に,完全相補的なホモ二本鎖DNA は分子量に依存した移動度を示します。しかしながら,一部にミスマッチが起きているヘテロ二本鎖DNAは,ミスマッチ部分における立体構造がホモ二本鎖 DNAとは異なるため泳動速度が遅くなります。ヘテロ二本鎖移動度分析は,この現象を利用して変異の有無を検出します。変異導入後の個体に対し,標的配列 の近傍領域に対するPCR を実施し,増幅産物を熱変性により乖離させ,再アニールを行うことでヘテロ二本鎖を形成させます。その後,試料を電気泳動することで,欠失や挿入の有無を 泳動パターンから識別することができます。
■ DNA/RNA分析用マイクロチップ電気泳動装置 MCE-202 MultiNA
MultiNAは,試料と専用試薬を装置にセットするだけで最大108試料まで自動分析ができる電気泳動装置です。また,サブマリン型電気泳動に比べ,同等のランニングコストで高い分離能と検出感度を有します。ヘテロ二本鎖移動度分析にMultiNAを活用することで,サブマリン型電気泳動では識別することが困難な短い欠失や挿入の有無を検出することができます。
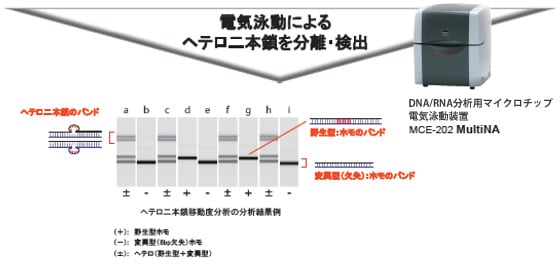
■ ヘテロ二本鎖移動度分析による変異導入した個体の選別例
F2個体を得るために,F1個体から同一変異を持つ雌雄ペアの選別を目的として,MultiNAを用いたヘテロ二本鎖移動度分析の例を紹介します。
飼育されたF1個体からゲノムDNAを抽出し,MultiNA によるヘテロ二本鎖移動度分析を行います。図に示すようにF1個体で複数のバンドパターンが観察されます。図の泳動事例では,野生型を除いて,A, B, C, Dの4パターンに分類できます。この同じパターンを示すF1個体の雌雄を選別して交配することにより,F2個体を得ます。
F2個体は,野生型,ヘテロ変異体,ホモ変異体が1:2:1の比率で出現することから,同様にヘテロ二本鎖移動度分析を行い選別されたホモ変異体の雌雄を交配することで,変異系統を確立することができます。
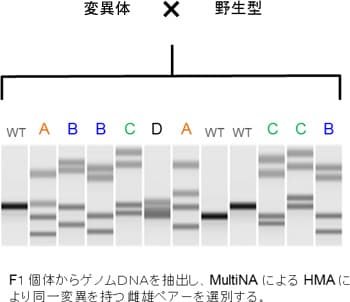
ヘテロ二本鎖移動度分析による同一変異を持つF1個体の選別
分析手法・データご提供:京都大学 農学研究科 木下 政人先生
関連情報
細胞培養ソリューション(クローニング)
細胞培養におけるクローニングでは,特定のDNA断片を持つ細胞系を検出・特定し,同じ遺伝子型をもつ細胞集団を作製すること(クローン株樹立)を目的とします。遺伝子導入・ゲノム編集細胞を対象としたクローン株樹立を効率化する新規システムをご紹介します。
関連製品

MCE-202 MultiNA
DNA/RNA分析用マイクロチップ電気泳動装置
ゲノム編集後の変異個体の評価や選別に最適
MultiNAを用いたヘテロ二本鎖移動度分析は,変異個体の評価や選別に有効です。野生型,ホモ変異体,ヘテロ二本鎖を移動度の違いにより区別することができます。
MultiNAは最大108検体までセットでき,しかも自動分析が行えます。多くのサンプルを分析する必要のある変異体のスクリーニングに活躍します。

Cell3iMager duos
ラベルフリーイメージングシステム
ゲノム編集後の細胞のクローニングにおけるシングルクローナルアッセイに最適
Cell3iMager duosは、ゲノム編集後の細胞のクローニングにおけるシングルクローナルアッセイに最適です。
独自の光学系を採用したラベルフリーイメージングシステムは、メニスカスの影響なくウェル全体の画像が取得可能ですので、細胞の見逃しがありません。
1細胞からの増殖の様子を確認できます。

Ampdirect® Plus
遺伝子増幅用試薬
ゲノム編集部位のPCRに最適
変異導入個体の識別には,個体組織からのゲノムDNA抽出,精製後,PCR増幅する必要があります。
Ampdirectは組織からのDNA抽出後に精製すること無く,PCRを実施することができるため,効率的にPCR増幅産物を得ることが出来ます。